
مقدمه: بیماریهای ذخیرهای دستهای مهم از بیماریهای ارثی اختلال متابولیسم هستند که بسته به مادهی تجمعی و محل تجمع، تظاهرات و عواقب و پیشآگهی متفاوت دارند. درگروهی از این بیماران تجمع پیشرونده گلیکوزآمینوگلیکان (موکوپلی ساکارید MPS) در لیزوزوم سلول ایجاد میشود. چهار ماده عمده در این بیماری درگیر هستند؛ تجمع هپارانسولفات بیشتر با مشکلات مغزی و تجمع کراتان سولفات، درماتان سولفات و کندروایتین سولفات با عوارض مزانشیمال همراه هستند. انواع شناخته شده و مهم این گروه انواع1، 2، 3، 4، 6 و 7 هستند (جدول1).
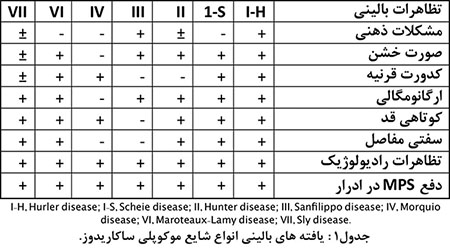
مشخصات بیماران: مشکل در نوع MPS-6 اختلال در آنزیم آریل سولفاتازB بهعلت موتاسیون در ژن ARSB واقع در بازوی بلند کروموزوم5 است. خصوصیات ظاهری معمولاً پساز یکسالگی خود را نشانداده و قبل از آن نسبتاً طبیعی هستند. بیماران این نوع تقریباً تمامی تظاهرات نوع یک این بیماری که شدیدترین این بیماران است را باشدت کمتر و سیر آهستهتر نشان میدهند.
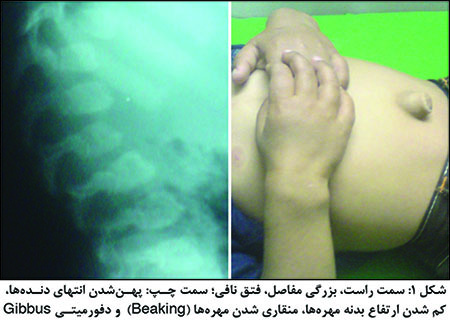
یافتههای مهم: کوتاهی قد، جمجمهی بزرگ، پُرمویی، کدورتقرنیه، صورت خشن و زبانبزرگ، مفاصل خشک و بزرگ، کبد و طحال بزرگ، کاردیومیوپاتی و بیماری دریچهای قلب (که میتواند علت مرگ زودرس این بیماران باشد). رادیوگرافی دست و مچ و طرفی ستون مهرهها (شکل1) تشخیص را مطرح کرده و با بررسی سطح آنزیم تشخیص قطعی میشود. تشخیص قبلاز تولد با یافتن موتاسیون در خانواده امکانپذیر است.
ثبت نظر